




▲第一作者:谢李燕;朱青;张国桢 ;通讯作者:江俊
通讯单位:中国科学技术大学化学与材料科学学院
论文DOI:10.1021/jacs.0c00561
全文速览
系统阐述了利用金属与金属氧化物功函数差异给后者注入电子从而诱导质子迁入氧化物晶格的物理化学机制,并发展了一套成熟的酸-金属联合处理应用方案,实现温和条件下金属氧化物半导体可控加氢和性质调控。
背景介绍
A加氢是调控材料性能最重要的技术之一
金属氧化物半导体是一类重要的半导体材料,具有丰富可调的电、光、磁学性质。金属氧化物氢化处理是调节其性质提升效用的重要方式。例如,氢化TiO2可增强光吸收改进光催化性能(Science 2011, 331, 746),氢化VO2会发生电子相变,拓宽其在光电领域的应用(Nat. Commun. 2018, 9, 818; Sci. Adv. 2019, 5, eaav6815),氢化WO3和MoO3可实现局域表面等离子共振的调控(J. Am. Chem. Soc. 2016, 138, 9316)。
B传统的加氢方法条件苛刻
传统的加氢策略用H2作为氢源,但因为H2键能高达436 KJ/mol,断开H-H键能耗很大,而且H原子进入金属氧化物半导体晶格也依然需要高能驱动,因此这个策略需要高温高压和贵金属催化剂,成本很高(Chem. Rev. 2019, 119 , 4777)。与此同时,酸溶液是虽然是方便而廉价的质子源,但利用酸来给金属氧化物加氢鲜有成功。
研究盲点:酸溶液中的氢质子为什么很少被用在金属氧化物半导体加氢上
研究出发点
利用酸溶液作为氢源,首先要了解利用酸质子给氧化物加氢的难点在哪。以往的实验观察到,金属氧化物在酸中要么表现为化学惰性(TiO2,WO3, MoO3和Nb2O5)要么会被酸腐蚀(如VO2。而且,直接用酸溶液实现金属氧化物的加氢需要解决加氢后电荷平衡的问题。这是因为阴离子体积大而无法进入金属氧化物晶格,仅有氢质子进入金属氧化物体相将破坏电中性,而且表面静电相斥作用也阻止氢质子在金属氧化物体相累积。我们从金属接触VO2可以阻止后者被酸腐蚀的实验现象(Nat. Commun. 2018, 9 , 818)出发,提出了一个大胆的设想,是否可以发展一套用金属辅助酸溶液中金属氧化物加氢的策略?于是,我们用第一性原理计算模拟了一批金属氧化物(TiO2,WO3, MoO3和Nb2O5)的金属-酸溶液联合加氢过程,随后做了实验对计算结果进行了验证。
我们的结论是,这个策略不仅可行,而且可以实现可控加氢(即调节氢化的程度)。其关键在于两方面的调控:一、金属与金属氧化物功函数差异;二、质子迁移进入金属氧化物晶格的势垒。我们随后对所得加氢产物的光,电,热,磁等性质变化进行了研究,发现加氢显著拓宽了金属氧化物材料性质的可变范围,为新材料的设计提供了理论指导。
图文解析
a. 机理图
这个工作的机理如图1所示主要分为三步:(1) 功函数的差异驱动电子在金属/金属氧化物界面处从金属流向金属氧化物,导致金属氧化物负电荷;(2)金属氧化物中的负电荷吸引溶液中的质子向晶格扩散;(3) 氢扩散到金属氧化物晶格后形成了稳定的氢掺杂。
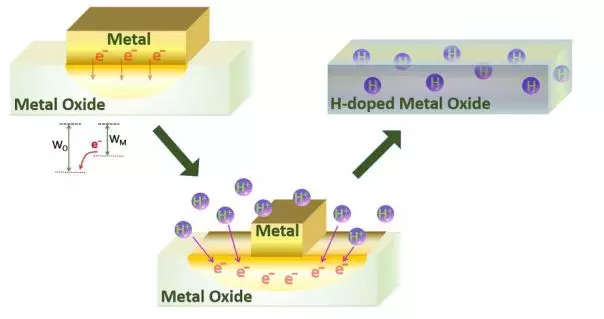
▲图1:金属诱导酸中氢质子加入金属氧化物体相实现金属氧化物加氢的机理图。
b. 理论计算部分。
首先,我们以锐钛矿型TiO2为模型体系来探讨这个加氢方法的可行性。我们选择了金属Al、Zn、Cu和Ag,它们的功函数(WF)分别是4.11、4.29、4.32和4.44 eV,均低于TiO2(6.67 eV),允许电子从金属流向TiO2。然后,我们对金属与TiO2(101)的界面模型来进行差分电荷分析,证实了电子在界面处从金属转移到TiO2(图2)。另外,Bader电荷分析显示这种界面电荷极化导致平均每个金属原子有0.02~0.09个负电荷转移到TiO2,使后者携带负电荷,而负电荷与酸溶液中质子的静电作用将变为氢原子进入晶体的驱动力。
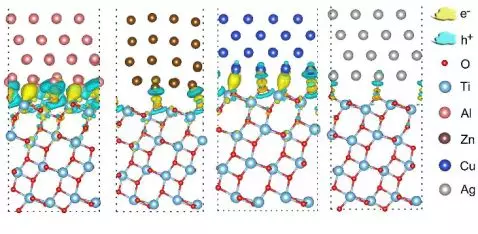
▲图2:电子从金属表面转移到金属氧化物表面示意图。黄色和蓝色分别代表电荷迁入和迁出的区域。
我们计算位于表面三种不同位置的氢原子迁入TiO2晶格的能垒,如图3所示。图3b-d中收集了不同电荷条件(1 h: 1个空穴, 0 e: 中性, 1 e: 1个电子)下质子迁移进其晶格的势能面。不论质子从哪个位置迁入,其势垒都随着体系负电荷的增加而降低,表明锐钛矿型TiO2中的负电荷累积可促进氢向其晶格的扩散。以从位点1开始迁移(图3b)为例,在中性条件(0 e)下,氢原子从晶格表面向次表面的迁移必须越过0.97 eV的势垒并吸收0.22ev的热量(ΔE)。相比之下,负电荷条件(1e)显著降低了能垒(0.47ev),并将吸热反应转变为放热反应(ΔE=-0.45ev),而正电荷条件(1h)能垒提高(1.24ev),反应吸热增加(ΔE=0.45ev)。注:ΔE=末态体系能量-初态体系能量。
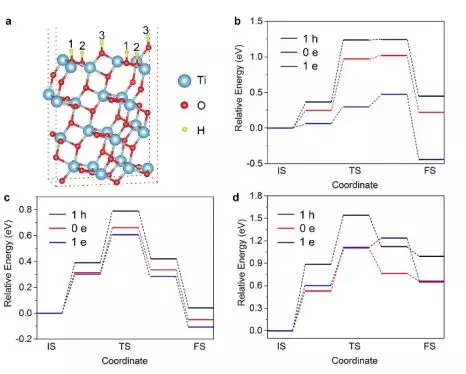
▲图3:电荷累积在金属氧化物上导致氢进入TiO2晶格的能垒降低。(a)H在锐钛矿型TiO2(101)表面的三个可能结合位点,标记为位点1/2/3。从位点1(b)、位点2(c)和位点3(d)开始的氢迁移进入TiO2晶格的能量变化图。起始位置的能量设置为0 eV。
c. 简单实验验证
作为理论研究为主的课题组,我们尝试进行简单实验,验证金属-酸溶液处理方法的氢化效果。用Zn金属与半导体混合,投入盐酸溶液中处理,白色的原始锐钛矿-TiO2迅速转变为蓝黑色(图4a)。图4b显示了1H NMR(核磁共振)光谱数据,指示TiO2晶格中的氢元素信号。如图4c所示,原始样品只吸收紫外光,而处理过的样品在可见光和红外区域显示出相当大的光吸收。ESR(电子自旋共振)谱在g=1.94处显示出强信号(图4d),这可归因于传输进来的电子大都局域在Ti原子附近,从而产生了Ti3+顺磁中心从而被ESR谱观察到。由于引入了Ti3+中心,掺杂H的TiO2样品显示出比原始TiO2更高的室温铁磁性(图4e)。并且理论计算也表明氢化破坏了电子自旋对称性,并且使费米能级升高实现金属-绝缘体转变(metal-insulator transition, MIT)。这将显著提高电子电荷载流子的浓度。进一步的电学实验证实,氢化后材料的电导率显著增加了5个数量级(图4f)。与此形成鲜明对比的是,高功函数的贵金属Au/Pd/Pt转移到TiO2的电荷要少得多。如所料,实验中的Au/Pt/Pd加酸处理也未能使TiO2氢化。
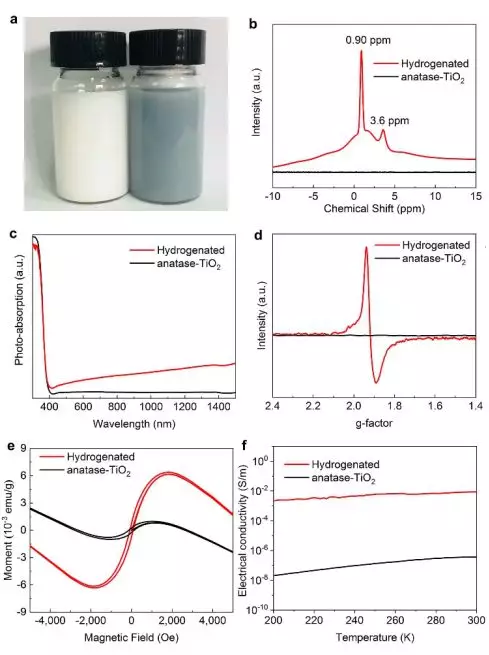
▲图4:利用Zn-盐酸处理实现TiO2加氢后材料的性能变化。原始和氢化锐钛矿型TiO2的(a)照片;(b)1H NMR谱;(c)UV-Vis-NIR吸收光谱;(d)ESR谱;(e)磁滞回线;(f)电导率随温度变化图。
d. 基于理论预测筛选金属与半导体,有效调控氢化浓度
我们接着用Zn-盐酸处理实现了Nb2O5,MoO3和WO3的加氢。第一性原理计算表明,这些金属氧化物具有比Zn更高的功函数,差分电荷分布图也表明电子会从Zn流入金属氧化物。如图5a所示,随着体系中负电荷的增加,H进入晶格体相的迁移势垒降低。 图5b-d 中H信号峰的出现和颜色变深均证实了这些氧化物被有效氢化。图5e表明加氢氧化物的可见光吸收增强。图5f所示,加氢导致氧化物电导率显著增加了4~7个数量级。计算还发现,质子迁移进ZrO2和SnO2体相的能垒非常高,因而不能用电子-质子共掺杂策略实现有效加氢。这一预测也得到了实验的证实。
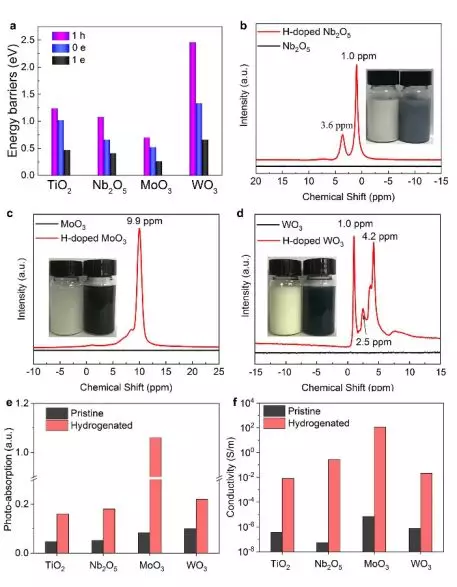
▲图5:Nb2O5, MoO3和WO3用Zn-盐酸方法实现加氢的情况。(a) 锐钛矿型TiO2(1位点)、Nb2O5, MoO3和WO3的H迁移入相应晶格的能垒变化图。(b)Nb2O5,(c)MoO3和(d)WO3的1H核磁共振谱,插图是锌-盐酸处理前后样品的照片。Zn-盐酸处理前后样品(e)700nm处的光吸收强度和(f)300K处的导电性。
第一性原理计算也发现,和Zn相比,Cu与金属氧化物的功函数差异更小,因此转移给氧化物的电荷较少。因此,Cu-盐酸处理的加氢过程会变慢(图5a),使金属氧化物加氢度易于调控。使H扩散势垒减少较少这使得我们可以很容易地调节H掺杂的程度。如图6a所示,Cu-盐酸处理实现了WO3的逐步加氢,控制掺氢浓度。图6b是我们模拟的不同程度WO3氢化物HxW8O24(x=0~4)的分元素轨道态密度图。费米能级(0 eV)以下的填充态(阴影区)是电荷载流子,表明氢化后的WO3有准金属特性。而且,该填充态内的电子数(0.88、1.78、2.72和3.60)随氢化度增加呈线性增大趋势,表明氢掺杂引起的载流子浓度变化可用氢掺杂量定量控制。
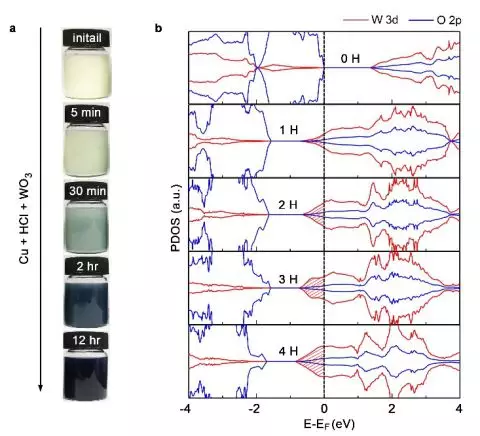
▲图6:WO3利用Cu-盐酸处理实现不同程度的加氢。(a)Cu-盐酸处理WO3后0-12小时的变化。(b)不同H掺杂浓度下WO3的分元素轨道态密度图(PDOS)。
总结与展望
在这项研究中,通过理论计算,我们确认金属氧化物氢化过程的热力学受控于金属-金属氧化物功函数差,动力学受控于从金属转移到金属氧化物的负电荷量。通过实验验证,我们在温和条件和酸溶液下实现了若干金属氧化物(锐钛矿型TiO2/WO3/MoO3/Nb2O5)的氢掺杂。而且,通过调节功函数差异,我们可以定量控制掺氢浓度,进而调控氢化氧化钨(HxWO3)的电子结构。我们预计,金属-酸联合处理法将成为金属氧化物可控加氢的一种通用策略,将助力光催化、光学器件和传感器等领域的新材料的理性设计。
免责声明:本网站所转载的文字、图片与视频资料版权归原创作者所有,如果涉及侵权,请第一时间联系本网删除。

官方微信
《中国腐蚀与防护网电子期刊》征订启事
- 投稿联系:编辑部
- 电话:010-62316606-806
- 邮箱:fsfhzy666@163.com
- 中国腐蚀与防护网官方QQ群:140808414

“海洋金属”——钛合金在舰船的

腐蚀与“海上丝绸之路”