
碳浓度对渗氮化合物层生长的影响
2022-08-09 16:04:10
作者: 每天学点热处理 来源: 每天学点热处理
分享至:


1 介绍
近年来,在提高汽车燃油经济性和静音性的背景下,要求变速器多级化和小型轻量化,零件需要进一步的高精度化和高强度化。最常用的表面硬化热处理是渗碳淬火,是利用奥氏体骤冷发生的马氏体转变的热处理方式,因此热处理导致零件变形是不可避免的。
氮化及软氮化,是将钢加热到450 ~ 600℃的铁素体区域,氨气分解反应而产生的N原子渗入钢的表面并扩散,形成Cr、Al等各种氮化物,是将N固溶在基体中从而使表面硬化的热处理方法[1]。由于这是不伴随相变的热处理,所以零件变形小,组装精度高,对提高汽车的静音性等很有效。对钢进行氮化处理后,在钢的最表层生成的铁氮化物(ε相:Fe2-3N, γ′相:Fe4N)等构成的化合物层。化合物层的变形比基体小,因此容易剥落,有时会成为接触面疲劳的破坏起点[2-3]。
近年来,随着氮化气氛的控制技术和组织分析技术的发展,越来越清楚组成化合物层的相的种类对疲劳强度的影响[4-6]。例如,小林等人对机械结构用碳钢JIS-S35C进行氮化处理,使材料的化合物层在气氛侧具有γ′相、基体侧具有ε相化合物层。该氮化材料的表面疲劳强度比化合物层以ε相为主体的传统氮化材料优越[4]。因此,为了实现氮化零件的高强度化,对化合物层相结构的控制是极为重要的课题。
Fe-N二元系中关于化合物层相结构控制的报告可以看到好几个。Fe-N二元体系中,在一般氮化条件下生成的相有ε相和γ′相两种。根据Fe-N二元系统平衡状态图[7]和Lehrer曲线图[8]可知,在N浓度高的区域ε相稳定,在N浓度低的区域γ′相稳定。因此,Fe-N二元体系中的化合物层的结构是γ′单相,或者是气氛侧为ε相、基体侧具有γ′相的两相。
也有一些关于Fe-C-N三元体系的化合物层控制的报告。例如,Slycke等报告了575℃时Fe-C-N三元体系平衡状态图[9, 10]。另外,Du等报告了Fe-C-N三元体系中关于Lehrer曲线图的计算[11, 12]。从这些报告可知,C是稳定ε相的元素。
另一方面,含有C的常用钢有时会呈现出与Fe-N二元体系不同的相结构。平冈等通过对机械结构用合金钢JIS-SCM435进行氮化,报告了与母相平衡的化合物层分别ε相和γ′相的情况[13]。此外,浅田等人用Fe-Cr-0.2C的合金钢为原料,研究了Cr的添加对化合物层的相结构的影响:随着Cr量的增加,与基体平衡的化合物层从γ′相变化为ε相为主体[14]。由于上述的平衡状态图很难解释这种差异,所以对控制化合物层相结构的氮化材料的推广应用造成了很大的阻碍。因此,弄清楚氮化过程中对化合物层的形成产生影响的各种因素,并就机理取得统一意见,可以说是在工业上极为重要的课题。
在常用钢中,与母相平衡的化合物层发生变化的原因之一,是热处理过程中钢表面脱碳对气氛的影响。脱碳是钢加热时不可避免的现象,据报道,在氮化处理中也会发生脱碳现象。因此,化合物层内的C浓度有可能随着时间而变化。因此,本研究通过对基体含碳量不同的钢进行氮化时表层合金元素的浓度分布以及相结构随时间的变化进行调查,探讨了Fe-C-N三元体系中产生各种相结构的机理。
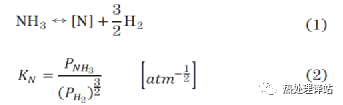

2实验方法
表1为试验材料的化学成分。这些是以一般机械结构用碳钢JIS-S30C为基础,使C含量变化为0、0.1、0.3及0.8%的成分系(以下称为C00、C10、C30、C80)。
表1 化学成分(mass%)

首先用真空熔炼炉熔制10kg的铸锭,然后通过热锻制成φ25mm的圆棒。接着,以JIS G0561为基准,将C00和C10以925℃加热,将C30以900℃加热,将C80以870℃加热30min。通过机械加工,将这些材料制成了10×50×2mm的试样,用于气体氮化处理。气体氮化处理使用NH3、N2气体,将试样在570℃保持0.5、1.0、3.0h后,在80℃的油中冷却。处理氛围为大气压力,由式(1)所示的NH3气体分解反应产生的H2分压(PH2),由热传导式H2传感器实时测量。另外,还使用泡泡瓶式NH3分析仪测量了NH3分压(PNH3)。以这些PH2和PNH3为基础,计算出式(2)所表示的氮势(以下称为KN)[16],并调整了导入炉内的NH3和N2气体的流量,使KN在目标值1.1 atm-1/2不变。每10分钟测量PH2、PNH3及KN的平均值,如表2所示。图1示出了由综合热力学计算系统Thermo-Calc2016b(热力学数据库:TCFEver.8.1, Fe- dataver .6)计算的(a) Fe-N二元体系和(b) Fe-0.8%C-N三元体系的Lehrer曲线图。作为比较,在图1 (a)中重叠实验Lehrer曲线图[8]。在本实验中,Fe- N二元系统(C00)在ε相和γ′相的边界附近,Fe-0.8%C-N三元系统(C80)在ε相区域进行了氮化。氮化处理后,用3%Nital(3%硝酸酒精溶液)腐蚀试样,在光学显微镜及扫描电子显微镜(SEM)观察断面表层部。另外,采用背散射电子光谱(EBSD)对化合物层的相进行了辨识。另外,采用辉光放电发光光谱分析装置(GDOES)对化合物层中的N、C、Si、Mn浓度进行了测定。
表2 渗氮条件



(a)Fe-N二元合金 (a)Fe-0.8%C-N三元合金
图1 计算的Lehrer图,与试验测量数据对比
3实验结果
3.1化合物层相结构随时间的变化
图2显示了氮化后试样断面表层的光学显微镜照片。在任何一种钢种及氮化时间下,在钢的表面都观察到被认为是化合物层的白色耐腐蚀层。化合物层的厚度随着母材的含C量和氮化时间的增加而增大。

图2 在570℃、KN = 1.1 atm-1/2条件下、时间分别为0.5、1.0和3.0h,用3% Nital腐蚀试样截面
图3显示了氮化后的C00表层的SEM图像以及EBSD法的相结构分析结果。氮化时间为0.5h时,在化合物层的深度方向表面侧形成了ε单相,在更深的位置形成了ε + γ′混相,在母相附近形成了γ′单相。即使氮化时间增加,该层叠顺序也不会改变,γ′单相区和ε单相区一起增长。

图3 0.0%C钢氮化0.5、1.0和3.0h后截面的SEM图像和EBSD相位影像
图4给出了氮化后的C10表层的SEM图像以及EBSD法的相结构分析结果。氮化时间为0.5h时,表层为ε单相,在更深的位置为ε + γ′混相。氮化时间1.0h、3.0h时,表层为ε单相,但在中间的位置ε相的比例下降,γ′相成为主体。再往里,虽然母相方面也以γ′相为主体,但与中间位置相比,存在较多的ε相。

图4 0.1%C钢氮化0.5、1.0和3.0h后截面的SEM图像和EBSD相位影像
图5显示了氮化后的C30表层的SEM图像以及EBSD法的相结构分析结果。氮化时间为0.5h时,整个化合物层是以ε相为主体的ε + γ′混相。氮化时间为1.0h和3.0h的产品与C10氮化材料一样,在中间位置以γ′相为主。另一方面,与C10相比,母相侧的ε相比例提高,化合物层的结构从表层到深度方向分别为ε单相、γ′单相、ε + γ′是混相。

图5 0.3%C钢氮化0.5、1.0和3.0h后截面的SEM图像和EBSD相位影像
图6显示了氮化后C80表层的SEM图像观察及EBSD法的相结构分析结果。在氮化时间1.0h之前,整个化合物层都是ε相主体的ε + γ′混相。氮化时间为3.0h的时候,呈现出与C30相同的结构,但深度方向中间位置的γ′相比例和母相一侧的ε相比例更高。

图6 0.8%C钢氮化0.5、1.0和3.0h后截面的SEM图像和EBSD相位影像
3.2化合物层中N、C浓度随时间的变化
图7表示对C00、C10、C30和C80进行氮化时N浓度曲线随时间的变化。N浓度在任何一种氮化时间内,都是最表面最高,之后随着离表面距离的增加而减少,在相当于化合物层厚度的深度后急剧下降。N浓度方面,最表面约为7.5mass%,深度中间位置约为6.5mass%,急速下降前约为5.7mass%,不论氮化时间长短,几乎都是恒定的。

图7 用GDOES测定的钢渗氮0.5、1.0和3.0h后N浓度分布
图8给出了C00、C10、C30和C80进行氮化时C浓度曲线随时间的变化。在C10、C30以及C80中,在化合物层的母相侧,可以看到C浓度高于母相的区域。据推测,这是C从母相到化合物层的浓化现象[17]。随着氮化时间的增大,C浓度的峰值呈现出降低的趋势。而且在最表面C浓度比母相低。这表明在氮化过程中,产生了从化合物层到气氛中的脱碳。随着氮化时间的延长,脱碳引起的C浓度下降量也越来越大。

图8 用GDOES测定的钢渗氮0.5、1.0和3.0h后C浓度分布
图9显示了对C80进行氮化时Si及Mn浓度曲线的时间变化。每种元素在深度方向上都是均匀的,没有向化合物层分配。由此可见,化合物层是以无再分配局部平衡或帕拉平衡增长的。

图9 用GDOES测定的0.8%C钢渗氮0.5、1.0和3.0h后Si和Mn浓度分布
4讨论
4.1化合物层的相结构与N、C浓度变化的对应关系
从Fe-C-N三元体系平衡状态图和计算的Lehrer曲线图可以看出,随着化合物层中的C浓度升高,ε相趋于稳定。另外,如果在化合物层和母相的界面上没有发现置换型元素的分配,那么预计化合物层的相结构随时间的变化会受到间隙型元素的强烈影响。因此,为了验证仅通过N、C浓度分布随时间的变化是否能够说明相结构随时间的变化,根据深度方向上N和C浓度的实测值计算出稳定相,并尝试与实际得到的化合物层相结构进行比较。计算采用Thermo-Calc2016b(热力学数据库:TCFEver.8.1),作为代表使用了C80的N和C浓度实测值。没有考虑Si和Mn,而是假设Fe-C-N三元体系中的稳定相为完全平衡进行了计算。
图10给出了根据氮化后的C80中的N、C浓度实测值通过平衡计算求出的ε相体积比例,以及与根据SEM-EBSD分析结果求出的ε相体积比例的比较。在任何一种氮化时间下,根据N、C浓度热力学计算出的ε相体积比例,与根据SEM-EBSD分析结果求出的ε相体积比例在精度上都很一致。这一事实支持了化合物层内的相结构随时间变化的原因是化合物层内N、C浓度分布随时间变化的推测。

图10 用EBSD法测定的渗氮0.5、1.0和3.0小时的0.8%C钢化合物层中ε相的体积分数分布,并根据N和C的分布,用Thermo- calc 2016b计算了ε相的体积分数分布
4.2氮化中表层C浓度随时间的变化
本节考察了母相C浓度对化合物层中C浓度随时间变化的影响。
图11给出了根据氮化后的C80中N、C浓度的实测值,通过平衡计算求出的N、C的化学势μN、μC的曲线。在任何一种氮化时间下,接触氮化气氛的钢表面的μN和μC几乎是恒定的。另外,钢表面的μN比钢内部的高,钢表面的μC比钢内部的低。另外,由于化合物层的厚度随着氮化时间的推移而增大,因此化合物层厚度方向上μN、μC的梯度随着氮化时间的推移而减小。由此可见,化合物层厚度方向上N、C浓度变化的驱动力随着氮化时间的推移而减小。

图11 根据N和C的分布,用thermocalc 2016b计算0.8%C钢氮化0.5、1.0和3.0小时化合物层中化学势分布
接着,在上述前提下,考察各种氮化时间下C浓度的时间变化与相结构的关系。
当氮化时间为0.5h时,在C10、C30、C80母相与化合物层的界面处,化合物层一侧的C浓度高于母相。据推测,这是由于化合物层厚度方向上C浓度变化的驱动力较大,C从母相向化合物层的浓化明显的结果。此外,即使在脱碳影响最大的化合物层表面附近,也比母相具有较高的C浓度,其值约为0.5mass%以上。其原因之一是C在化合物层及母相中的扩散速度的差异[18]。在化合物层内,C的扩散速度明显小于母相,因此可以认为0.5h氮化后的化合物层受C从母相浓化的影响几乎变成了ε单相。
当氮化时间为1.0h时,C10、C30的化合物层中间附近的C浓度下降到0.2 ~ 0.3mass%以下。其原因是,化合物层越厚,厚度方向的μC的梯度变小,除了母相供给的C量下降外,而且,此前从母相浓化来的C在化合物层中被稀释了,这可能是因为从化合物层向气氛中的脱碳的影响,比0.5 h的氮化材料相对显著了。结果,在中间附近N浓度降低,C浓度也降低,γ′相趋于稳定。C80中由于母相的C浓度高,向化合物层的C供给量大,所以中间附近的C浓度较高,约为0.6mass%。因此,即使氮化时间达到1.0h,整个化合物层中ε相仍然稳定存在。
如果氮化时间达到3.0h,化合物层进一步增厚,即使母相C浓度达到C80,对化合物层的C供给量也会下降,相对来说脱碳的影响会变得显著。因此,化合物层一侧的C浓度下降到与母相相同,中间附近的C浓度也下降到0.3%左右。因此,可以认为在中央附近γ′相稳定化了。
综上所述,我们明确了化合物层的相结构随时间变化的主要因素是化合物层中厚度方向上的C浓度随时间变化。进一步表明,化合物层中C浓度随时间的变化是由于C从母相向化合物层的浓化以及从化合物层表面附近向气氛中的脱碳受到厚度方向上μC梯度变化的影响而产生的。
5 结论
为了明确C浓度变化对氮化钢表层化合物层生长的影响,调查了母相C浓度不同的钢在氮化时表层合金元素的浓度分布以及相结构随时间的变化。得到的结论如下:
1)在母材C量为0.1mass%以上的钢中,氮化时间仅为0.5h的情况下,化合物层结构以ε相为主。另一方面,随着氮化时间延长到1.0h和3.0h,化合物层厚度方向中间附近γ′相的比例也随之提高。
2)无论在哪个时间,表层的Si和Mn浓度在深度方向上都是均匀的,没有向化合物层分配。
3)化合物层内相结构随时间变化的主要因素是化合物层内C浓度分布随时间变化。
4) C浓度分布的时间变化是由于C从母相向化合物层的浓化、从化合物层表面附近向气氛中的脱炭受到厚度方向上μC的梯度变化的影响而产生的。
免责声明:本网站所转载的文字、图片与视频资料版权归原创作者所有,如果涉及侵权,请第一时间联系本网删除。
相关文章

官方微信
《腐蚀与防护网电子期刊》征订启事
- 投稿联系:编辑部
- 电话:010-62316606-806
- 邮箱:fsfhzy666@163.com
- 腐蚀与防护网官方QQ群:140808414
点击排行
PPT新闻

“海洋金属”——钛合金在舰船的
点击数:8158

腐蚀与“海上丝绸之路”
点击数:6488