
2021年第一篇《Nature》封面文章诞生!
2021-01-08 10:49:44
作者:材料科学与工程 来源:材料科学与工程
分享至:



结构无序的材料存在一些基本问题,包括不同无序相(多晶硅)如何共存以及如何从一种相转变为另一种相。目前,非晶态硅得到了广泛研究;它在环境条件下形成四倍配位的共价网络,在压力下形成更高配位的金属相。然而,由于最先进的实验和计算技术的内在局限性(例如,通过模拟可以获得的系统尺寸),对无序硅结构转变的详细机理一直缺乏理解。
近日,来自英国牛津大学的VolkerL. Deringer等研究者,展示了如何在精确的量子力学计算上训练原子机器学习模型,以此可用来描述10万个原子(10 nm长度尺度)体系的液态非晶和非晶-非晶转变,预测结构、稳定性和电子性质。相关论文以题为“Origins of structural and electronic transitions in disordered silicon”发表在国际顶级期刊Nature上。同期,该文章登上了2021年第一期《Nature》的封面。
论文链接: https://www.nature.com/articles/s41586-020-03072-z

在理解结构复杂的材料,如液体和非晶态物质方面,计算机模拟在很大程度上已经达到了最先进的水平。然而,无序相对模拟提出了持续的挑战,需要大的系统尺寸,长时间的模拟和可转移的原子相互作用模型(即,对所有相关的结构和键合环境都有效的模型)。机器学习驱动的原子间势,是解决这些挑战的一种新兴且强大的方法;硅晶体相之间的压力诱导转变是这些方法最早的应用之一,最近的应用包括液相中的晶体成核。研究者之前基于分子动力学模拟,使用量子精确高斯近似势(GAP)机器学习模型,使用512到4096个原子之间的系统大小,并且只考虑当时的环境压力状态,对无序硅进行了初步研究。
在此,研究者利用更广泛的间隙分子动力学(GAP-MD),模拟了一个包含100,000个硅原子的系统,以解决各种结构跃迁的原子机制——包括那些在非常高的压力和密度下。在这个系统大小下,这种模拟包含了几百万个单独的时间步长,在此之前,这种模拟只能在(必然)有限的精度和可转移性的经验参数化力场下才能实现。研究者证明了此前简单的力场无法再现硅中由压力引起的变化,而这些变化在实验中已经观察到,在本研究中也发现了。
研究者的模拟揭示了非晶硅在增加外部压力下的三步转序列。首先,多晶低-、高密度非晶区并存,而不是依次出现。然后,观察到一个结构坍塌成一个明显的高密度非晶(VHDA)相。最后,研究者的模拟表明了这种VHDA相的瞬态性质:它会迅速成核,最终导致多晶结构的形成,这与实验一致,但在早期的模拟中没有看到。电子态密度的机器学习模型证实了:金属丰度在VHDA形成和随后的结晶过程中的开始。这些结果阐明了硅的液态和非晶态,并且在更广泛的背景下,它们例证了一种机器学习驱动的方法来预测材料建模。
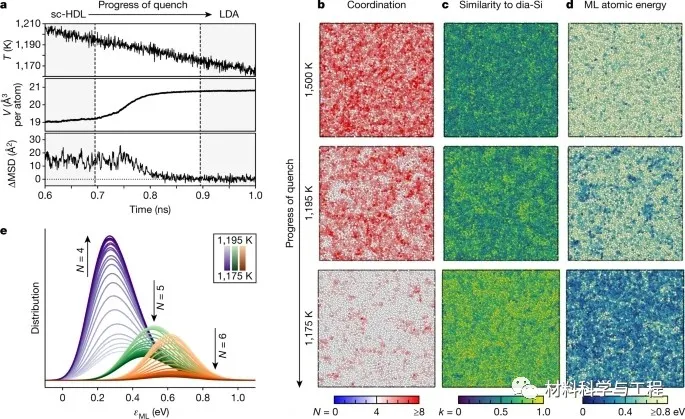
图1 过冷液态硅的玻璃化。
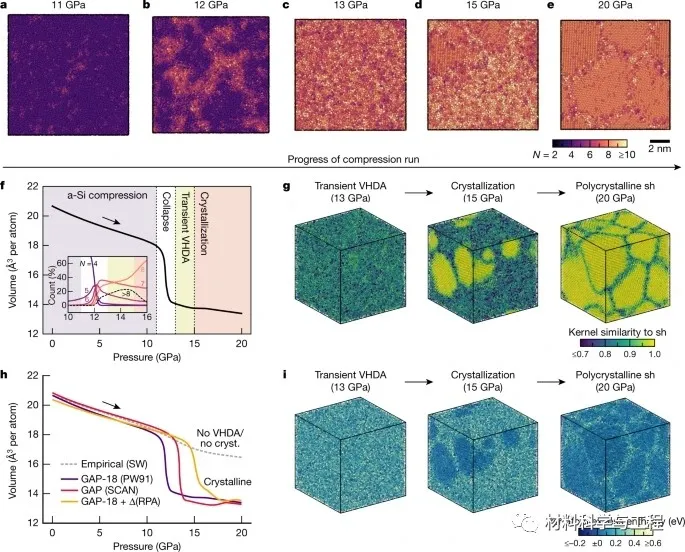
图2高压和超高压下的非硅。
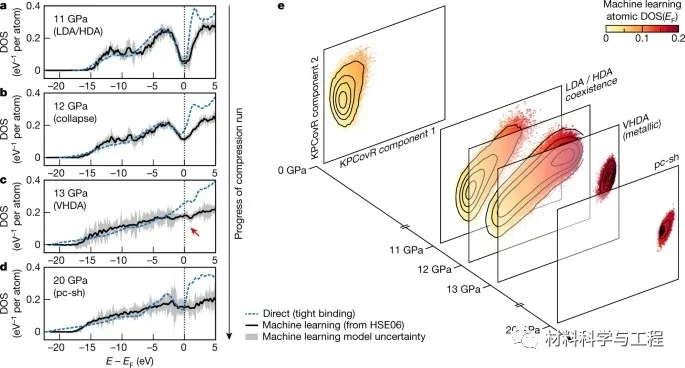
图3 结构转变的电子指纹。
综上所述,研究者模拟已经描述和解释了无序硅的全部相变,直到确定的极限(即结晶),与实验观察一致。然而,除了这一特定材料之外,目前的结果表明,原子机器学习方法可以引导科学发现。这些方法提供了对结构、稳定性和性质的量子精确预测,可以揭示迄今未知的现象:单个原子的结构和电子指纹,也可以揭示多晶、多晶和其他形式的纳米尺度不均一性。因此,对无序材料的模拟迈出了定性的一步:从简单的结构模型到在实验具有挑战性的条件下对材料系统的现实的、可预测的和完全原子的描述。
免责声明:本网站所转载的文字、图片与视频资料版权归原创作者所有,如果涉及侵权,请第一时间联系本网删除。
相关文章

官方微信
《中国腐蚀与防护网电子期刊》征订启事
- 投稿联系:编辑部
- 电话:010-62316606-806
- 邮箱:fsfhzy666@163.com
- 中国腐蚀与防护网官方QQ群:140808414
点击排行
PPT新闻

“海洋金属”——钛合金在舰船的
点击数:7130

腐蚀与“海上丝绸之路”
点击数:5741